


Chemicals
Activated carbon fabric ACC-5092-10 cloth was purchased from Kynol. The cloth was activated for 1 h at 100 °C in a vacuum oven before use. Potassium hydroxide (99%), potassium bicarbonate (99%) and sodium hydroxide (99%) were purchased from Sigma-Aldrich. All chemicals were of analytical grade and directly used as received without further purification.
Electrochemistry
All electrochemical measurements were performed at room temperature using a BioLogic SP-150 potentiostat and a Biologic BCS-800 Series. A coiled platinum wire (BASi, catalogue no. MW-1033) was used as a counter electrode. In three-electrode measurements, the reference electrode was Hg/HgO (ALS, catalogue no. RE-61AP) with 0.1 M KOH filling solution; the filling solution was exchanged routinely to keep the potential constant. Potentials were converted to the SHE using the correction ESHE = EHg/HgO + 0.165 V.
Elemental analysis
C, H and N wt% were determined by means of CHN combustion analysis using an Exeter Analytical CE-440, with combustion at 975 °C.
Volumetric gas sorption measurements
N2 isotherms were collected using an Autosorb iQ gas adsorption analyser at 77 K. The BET surface area was determined by the BET equation and Rouquerol’s consistency criteria implemented in AsiQwin. All pore size distribution fittings were conducted in AsiQwin using N2 at 77 K on carbon (slit-shaped pores) quenched solid density functional theory model. CO2 sorption isotherms were also collected on an Autosorb iQ gas adsorption analyser. Isotherms conducted at 25, 35 and 45 °C were measured using a circulating water bath. Samples were activated at 100 °C in vacuum for 15 h before gas sorption measurements.
Thermogravimetric gas sorption measurements
Thermogravimetric CO2 adsorption experiments were conducted with a flow rate of 60 ml min−1 using a TA Instruments TGA Q5000 equipped with a Blending Gas Delivery Module. Samples were activated under flowing N2 for 30 min at various temperatures before cooling to 30 °C and switching the gas stream to CO2 mixtures. Cycling experiments were carried out on a Mettler Toledo TGA/DSC 2 Stared system equipped with a Huber mini chiller. For tests with high-concentration CO2, the adsorption and desorption of CO2 were performed at 30 and 100 °C for 20 min under 30% CO2 and 70% N2 with a flow rate of 140 ml min−1, respectively. For DAC tests, adsorption was carried out at 30 °C for 60 min, with 400 ppm CO2 in dry air, and desorption was carried out at 130 °C for 60 min with 100% N2.
Adsorption microcalorimetry measurements
The simultaneous measurement of the heat of adsorption and the adsorbed amount of carbon dioxide was performed by means of a heat flow microcalorimeter (Calvet C80 by Setaram), connected to a high-vacuum (residual pressure less than 10−4 mbar) glass line equipped with a Varian Ceramicell 0–100 mbar gauge and a Leybold Ceramicell 0–1,000 mbar gauge. Before the measurement, both PCS-OH and blank carbon cloth (roughly 150 mg before activation) were activated for 24 h under high vacuum (residual pressure less than 10−3 mbar) at 100 °C (temperature ramp 3 °C min−1). The adsorption microcalorimetry measurements were performed at 30 °C by following a well-established step-by-step procedure described in detail elsewhere34. This procedure allowed, during the same experiment, for the determination of both integral heats evolved (−Qint) and adsorbed amounts (na) for small increments of the adsorptive pressure. The partial molar heats obtained for each small dose of gas admitted over the sample were computed by applying the following ratio: ΔQint/Δna, kJ mol−1. The (differential) heats of adsorption are then reported as a function of CO2 adsorbed amount, to obtain the (differential) enthalpy changes associated with the proceeding adsorption process. The equilibration time in the microcalorimetric measurement was set to 24 h for small equilibrium pressures (less than 30 mbar), whereas it was reduced to 2 h for larger doses for PCS-OH. The equilibration time was reduced to 2 h (regardless of the equilibrium pressure) for the bare carbon cloth, as equilibration is expected to occur faster in absence of specific adsorption sites.
X-ray diffraction
Powder X-ray diffraction patterns were collected on a Malvern Panalytical Empyrean instrument equipped with an X’celerator Scientific detector using a non-monochromated Cu Kα source (λ = 1.5406 Å). The data were collected at room temperature over a 2θ range of 3–80°, with an effective step size of 0.017°.
Scanning electron microscopy (SEM)
Samples were mounted onto a stainless-steel SEM stub using adhesive carbon tape. SEM imaging was performed on a Tescan MIRA3 FEG-SEM. Analysis of SEM images was conducted using the FIJI ImageJ software.
NMR spectroscopy
Solid-state NMR experiments were performed with a Bruker Advance spectrometer operating at a magnetic field strength of 9.4 T, corresponding to a 1H Larmor frequency of 400.1 MHz. A Bruker 4 mm HX double resonance probe was used in all cases. 1H NMR spectra were referenced relative to neat adamantane (C10H16) at 1.9 ppm and 13C NMR spectra were referenced relative to neat adamantane (C10H16) at 38.5 ppm (left-hand resonance). All the NMR tests were conducted with a sample magic angle spinning rate of 12.5 kHz. A 90° pulse-acquire sequence was used in each experiment. For 13C NMR experiments, recycle delays were set to be more than five times the spin-lattice relaxation time for each sample to ensure that the experiments were quantitative.
Charged-sorbents with different water contents were prepared for the NMR characterization. The sorbents were kept in a closed container for 24 h under different RHs. Saturated Mg(NO3)2 solutions were used to maintain 53% RH at 25 °C, respectively28.
Titration measurements
First, 88 mg of sample was immersed in 2 ml of deionized water and sonicated for 20 min at 25 °C. The pH value was then recorded with a pH meter (Insmark IS128C, calibrated with buffer solutions before use) at 25 °C as the initial point. Second, 100 µl of HCl (0.1 M) was slowly added. The mixture was sonicated for 20 min at a constant 25 °C and the pH of the solution was recorded. The second step was repeated until the end of the titration. There was no weight loss due to evaporation during the titration.
13CO2 dosing for solid-state NMR experiments
Freshly activated samples (75 °C, vacuum oven, 24 h) were packed into 4 mm NMR rotors in air and then evacuated for a minimum of 10 min in a home-built gas manifold29. 13C-enriched CO2 gas (Sigma-Aldrich, less than 3 atom% 18O, 99.0 atom% 13C) was then used to dose the samples with gas at room temperature until the gas pressure stabilized, before the rotors were sealed inside the gas manifold with a mechanical plunger.
DAC tests in sealed chambers
The DAC tests were carried out in a sealed box (volume roughly 600 ml) with a CO2 sensor (Aranet4) to record the concentration of CO2, temperature and RH at every 1 min interval. Before each cycle, the box was exposed to fresh air until the CO2 concentration, RH and temperature stabilized. The sorbent was then placed in the box, which was sealed during measurements.
Joule heating
After each DAC adsorption step, the sorbent was extracted from the box and connected with an external power source for Joule heating. A BioLogic SP-150 potentiostat was used to vary the electrical input. A constant voltage was applied and adjusted during the experiment to achieve a sample temperature in a range of 85–95 °C under an N2 atmosphere. The temperature was monitored using a thermocouple at a single contact point. After Joule heating regeneration, the electrode was reused for another DAC adsorption cycle.
Preparation of charged-sorbents
Three-electrode method
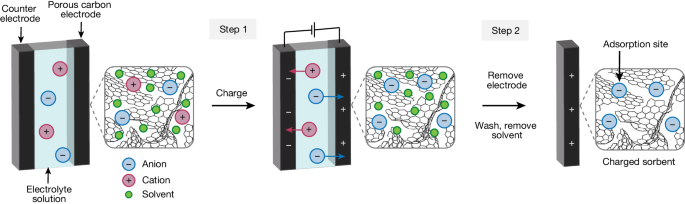
The charged-sorbents were prepared in a three-electrode configuration with a home-made cell, a Hg/HgO (in 0.1 M KOH) reference electrode and a platinum wire counter electrode. The activated carbon fabric ACC-5092-10 cloth (2 × 2 cm) was charged with a constant potential for 4 h (0.565 V versus SHE for the PCS-OH and −0.235 V versus SHE for NCS-K, respectively) in 40 ml of 6 M KOH(aq.) by means of chronoamperometry (Extended Data Fig. 2). After completing the charging process, the charged cloth was removed and held by plastic tweezers and rinsed with deionized water from a wash bottle for 5 min in total on both sides. Next, 500 ml deionized water in total was used to wash off the residual KOH solution. The rinsed cloth was then placed in the vacuum oven at 75 °C for 24 h to remove the remaining water.
For the soaked control sample, the activated carbon fabric ACC-5092-10 cloth (2 × 2 cm) was soaked in 40 ml of 6 M KOH for 4 h. After soaking, the same rinsing and drying processes used for the charged-sorbents were carried out. As a further control, a dripped cloth sample was prepared by adapting a literature protocol13. Here, 200 μl of 6 M KOH was dripped onto the surface of 340.0 mg ACC-10. Subsequently, the dripped samples were left in a Schlenk flask connected to a vacuum to let the samples dry for 72 h at room temperature.
Two-electrode Swagelok method
Free-standing carbon films were prepared by adapting the published method in ref. 35. In brief, YP80F activated carbon powder (95 wt%) (Kuraray Chemical) was mixed with polytetrafluoroethylene binder (5 wt%) (Sigma-Aldrich, 60 wt% dispersion in water) in ethanol. The resulting slurry was kneaded and rolled to give a carbon film of roughly 0.25 mm thickness, followed by removing residual solvent at 100 °C in vacuum for at least 24 h. Disc-shaped electrodes were then cut from the carbon films using a 0.25 inch hole punch. Symmetrical Swagelok electrochemical cells were then prepared in Swagelok PFA-820-6 fittings with stainless-steel current collectors, YP80F film electrodes (for both the positive and negative electrodes), 6 M KOH(aq.) electrolyte and a glass fibre separator (Whatman glass microfibre filter (GF/A)). Cells were charged at a constant cell voltage of 0.8 V for 4 h in two-electrode mode, and the positive electrode was then extracted, washed and dried, as above, to yield a charged-sorbent referred to as PCS-OH (YP80F). Three samples from three independent electrochemical cells were combined to provide sufficient material for gas sorption measurements.




{kind=link}
{kind=link}
{kind=link}
{kind=link}